Entwicklung und Barrierefunktion der Blutgefäßwand
Forschungsbericht (importiert) 2006 - Max-Planck-Institut für molekulare Biomedizin
Blutgefäße versorgen die Gewebe des Körpers mit Nahrungsstoffen und ermöglichen es den Leukozyten, Abwehrzellen des Immunsystems, im Organismus zu rezirkulieren. Da das Blutvolumen nur circa 5 bis 7% des Körpervolumens ausmacht, ist es für die Bekämpfung von Infektionen von entscheidender Bedeutung, dass Leukozyten in der Lage sind, aus dem Blut auszutreten und in Gewebe einzudringen. Dieser Vorgang leitet Entzündungsreaktionen ein und hält sie in Gang. Er stellt damit sicher, dass Infektionserreger in den verschiedenen Geweben des Körpers überhaupt durch Leukozyten erreicht und bekämpft werden können. Auch andere Schädigungen des Körpers durch mechanische oder chemische Reize lösen Entzündungen aus, locken Leukozyten an und leiten damit den komplexen Vorgang der Wundheilung ein.
Dies sind die positiven Aspekte des Entzündungsprozesses, die sicherstellen, dass der Organismus Infektionen überlebt und verwundetes Gewebe zumindest in Teilen regenerieren und heilen kann. Die negative Seite des Entzündungsprozesses kommt zum Tragen, wenn die Abwehrreaktionen der Leukozyten sich durch pathologische Regulationsmechanismen gegen körpereigenes Gewebe richten (Autoimmunerkrankungen) oder wenn eine akute und durchaus notwendige, hilfreiche Entzündungsreaktion aus verschiedenen (oftmals nicht genau verstandenen) Gründen nicht wieder abklingt, sondern chronisch wird. In diesen Fällen kommt es zur Zerstörung von Gewebe und zu nur schwer behandelbaren Krankheiten wie beispielsweise rheumatoider Arthritis, Parodontose oder multipler Sklerose.
Es ist also von großer medizinischer Relevanz, die molekularen Grundlagen zu verstehen, die den Eintritt von Leukozyten aus dem Blut in Gewebe kontrollieren. Aus den Forschungsarbeiten der letzten 15 Jahre ist bekannt, dass der Vorgang der Leukozyten-Extravasation (Austritt aus den Blutgefäßen) in mehreren Schritten erfolgt. Durch das kontrollierte Zusammenspiel verschiedener Klassen von Zelladhäsionsmolekülen und chemo-attraktiver Signalstoffe werden Leukozyten in entzündeten Geweben aus dem Blutstrom abgefangen und an die Oberfläche der Endothelzellen gebunden. Daran sind vor allem die Selektine beteiligt, eine kleine Gruppe von drei Adhäsionsmolekülen, die an bestimmten Kohlenhydratstrukturen auf bestimmten Trägerproteinen binden. Außerdem sind Integrine und die so genannten Chemokine beteiligt. Während die meisten dieser Moleküle bekannt sind, ist noch weitgehend ungeklärt, wie Leukozyten nach der Anheftung ans Endothel die Blutgefäßwand durchdringen, ein Prozess, der als Diapedese bezeichnet wird. Prinzipiell scheinen Leukozyten den Endothelzellverband auf zwei sehr unterschiedlichen Wegen zu überwinden: zum einen durch die geöffneten Kontakte zwischen den Endothelzellen hindurch und zum anderen sogar mitten durch den Körper von Endothelzellen. Es ist noch unbekannt, ob beide Wege gleichberechtigt benutzt werden. Allerdings sind die Mehrzahl der bisher bekannten endothelialen Zelloberflächenmoleküle, die am Prozess der Diapedese beteiligt sind, an den Kontakten zwischen Endothelzellen zu finden. Dies spricht dafür, dass Leukozyten hauptsächlich den Weg zwischen den Zellen hindurch benutzen.
Die Forschergruppe um Dietmar Vestweber hat in den letzten Jahren eine ganze Reihe von Glykoproteinen auf der Oberfläche von Endothelzellen und an Endothelzell-Kontakten identifiziert, die an der Auswanderung von Leukozyten aus den Blutgefäßen beteiligt sind. Ein Teil der Arbeitsgruppe widmet sich den Selektinen, ihren Liganden und einem humangenetischen Defekt, der die Biosynthese dieser Liganden betrifft; er wird Leukozyten-Adhäsions-Defizienz-II (LAD-II) genannt.
Ein größerer Teil der Gruppe konzentriert sich zurzeit darauf, molekulare Mechanismen zu verstehen, die die Öffnung und Bildung endothelialer Zellkontakte kontrollieren. Das Verständnis dieser Mechanismen wird es zum einen ermöglichen, die Einwanderung von Leukozyten in Gewebe zu beeinflussen. Zum anderen stellt die Ausbildung endothelialer Zellkontakte einen wesentlichen Schritt bei der Entwicklung des Blutgefäßsystems, der Angiogenese, dar.
Verschiedene molekulare Defekte, die zur Leukozyten-Adhäsions-Defizienz-II führen
Bei LAD-II handelt es sich um einen bis vor kurzem noch ungeklärten humanen Gendefekt, der zum völligen Fehlen fukosylierter Glykokonjugate führt. Dies impliziert auch das Fehlen der Selektinliganden. Diese Krankheit prägt sich zum einen, weil Selektinliganden fehlen, als Immundefizienz aus, die zu ständigen Infektionen und Fieberschüben und zu einem dramatischen Anstieg der Leukozytenzahlen im Blut führt. Zum anderen kommt es zu deutlichen psychomotorischen Entwicklungsstörungen und Defekten höherer neuronaler Funktionen. In Zusammenarbeit mit der Universitäts-Kinderklinik in Münster konnte jüngst eine Therapie des immunologischen Teils dieser Krankheit entwickelt werden, indem oral die Vorläufersubstanz der fehlenden Selektinliganden-Zuckerstruktur, die Fucose, gegeben wurde.
Mithilfe der kultivierten Zellen eines Patienten war es der Gruppe von Dietmar Vestweber vor wenigen Jahren möglich, den Gendefekt der Fukosylierungsdefizienz aufzuklären. Das
defekte Gen wurde kloniert und es stellte sich heraus, dass es für einen GDP-Fucose-Transporter kodierte. Die Ursache der Erbkrankheit LAD-II liegt also in der fehlenden Bereitstellung von GDP-Fucose im Golgi, einem für die Glykosylierung von Glykokonjugaten essentiellen Zellorganell.
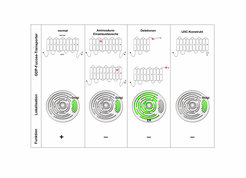
In der Zwischenzeit konnte diese seltene Krankheit bei weiteren Patienten diagnostiziert werden. Die genaue Untersuchung der defekten GDP-Fucose-Transporter zeigte, dass es verschiedene Typen von Mutationen gab. Weitere Untersuchungen erlaubten die Identifizierung struktureller Abschnitte des Transporters mit unterschiedlichen Funktionen. Bestimmte Abschnitte sind notwendig für die Lokalisation des Transporters im Golgi, andere für seine Transportfunktion (Abb. 1) [1,2].
 und solche, die zusätzlich noch die Lokalisation des Transporters im Golgi, einem für Glykosylierungen unerlässlichen Zellorganell, beeinträchtigen (rechts). Links ist der normale GDP-Fucose-Transporter dargestellt. Die Sequenz des Transporters enthält zehn putative Transmembran-Domänen, die als Kästchen dargestellt sind. Dieses Bild wurde im FEBS Journal publiziert [2].")
Ein endotheliales Membranprotein, das Diapedese und endotheliale Permeabilität beeinflusst
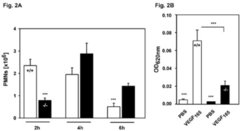
Vor wenigen Jahren hat die Arbeitsgruppe ein endotheliales Membranprotein entdeckt, genannt „Endothelial Specific Adhesion Molecule“ (ESAM), das selektiv an so genannten tight junctions exprimiert wird. Dies sind Kontaktstrukturen, die die Polarität von Endothelzellen sowie die Permeabilitätsbarriere der Gefäßwand aufrechterhalten. Durch die Erzeugung von Mäusen, denen das Gen für ESAM fehlt, konnten die Wissenschaftler zeigen, dass ESAM an einem frühen Schritt der Einwanderung von Leukozyten in entzündetes Gewebe beteiligt ist (Abb. 2). Gleichzeitig fanden sie, dass ESAM ganz allgemein in die Öffnung endothelialer Zellkontakte involviert ist. Bestimmte Substanzen, die die Durchlässigkeit der Gefäßwand für lösliche Bestandteile des Blutes steigern, bewirkten in ESAM-defizienten Mäusen eine deutlich geringere Permeabilitätssteigerung (Abb. 2). Damit wurde erstmals gezeigt, dass ein für die Öffnung von Endothelkontakten verantwortliches Membranprotein ebenfalls eine Rolle spielt beim Austritt von Leukozyten. ESAM stellt also einen idealen Ansatzpunkt zur Untersuchung von Mechanismen dar, die vaskuläre Permeabilität und Leukozytenaustritt regulieren.
 In normalen Mäusen (+/+) und in ESAM-defizienten Tieren (-/-) wurde die Einwanderung von Leukozyten in den entzündeten Bauchraum gemessen. Eingedrungene Leukozyten wurden 2, 4 und 6 Stunden nach Induktion der Entzündung gezählt. Nach 2 Stunden waren in ESAM-defizienten Tieren deutlich weniger Leukozyten eingewandert als in genetisch unveränderten Mäusen. Zu späteren Zeitpunkten erholte sich das System von dem Defekt. B) Genetisch unveränderten Mäusen (+/+) und ESAM-defizienten Mäusen (-/-) wurde zunächst intravenös ein Farbstoff und 10 Minuten später in die Haut der Wachstumsfaktor VEGF injiziert. Als Kontrolle wurde der Puffer PBS injiziert. 30 min später wurde der Austritt des Farbstoffs aus den Gefäßen in das umliegende Hautgewebe gemessen. Es zeigte sich, dass die durch VEGF ausgelöste Permeabilitätssteigerung in ESAM-defizienten Mäusen deutlich schwächer ausfiel als in genetisch unveränderten Tieren. Diese Ergebnisse wurden im Journal of Experimental Medicine publiziert [3].")
Mechanismen, die die Adhäsionsfunktion von VE-cadherin beeinflussen
VE-cadherin ist das wichtigtse Adhäsionsmolekül, das den Zusammenhalt endothelialer Zellkontakte vermittelt. Während ESAM eher regulatorische Funktionen bei der Öffnung von Endothelkontakten wahrnimmt (s.o.) und das Fehlen von ESAM selbst die Bildung des Blutgefäßsystems und die Stabilität endothelialer Kontakte nicht beeinträchtigt, führt das Fehlen von VE-cadherin in gendefizienten Mäusen dazu, dass die Bildung des Blutgefäßsystems während der Embryonalentwicklung gestört wird. Mäuse mit einer Gendefizienz im VE-cadherin-Gen sind daher nicht lebensfähig.
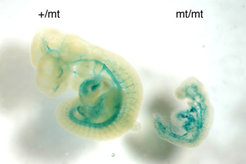
Die Forschergruppe um Dietmar Vestweber hat kürzlich gefunden, dass eine Endothel-spezifische Phosphatase (VE-PTP) mit VE-Cadherin interagiert und die Adhäsionsfunktion von VE-cadherin dabei wesentlich unterstützt. Die Untersuchung von gendefizienten Mäusen ergab, dass VE-PTP ebenso wie VE-cadherin für die Aufrechterhaltung und Remodellierung eines primitiven vaskulären Netzwerkes während der Embryonalentwicklung unverzichtbar sind. Die Defizienz des VE-PTP-Gens führt zu embryonaler Letalität und zur Zerstörung des Systems der Blutgefäße (Abb. 3). Eine normale Hierarchie von großen und kleineren Gefäßen konnte sich nicht ausbilden. Außerdem verblieben Endothelzellen in Gewebeexplantaten der Embryonen dieser gendefizienten Mäuse nicht im Gefäßverband, sondern verließen diese Strukturen und wuchsen als Zellschichten weiter. Andere Phosphatasen, die ebenfalls mit VE-cadherin und damit komplexierten intrazellulären Proteinen assoziieren, sind nicht essentiell für die embryonale Angiogenese. Damit kommt VE-PTP warscheinlich eine wesentliche Rolle als Regulator endothelialer Kontakte zu. Derzeit wird untersucht, ob VE-PTP an der Diapedese von Leukozyten beteiligt ist.
 und einem VE-PTP-defizienten Embryo (rechts), beide im Embryonalstadium E10.0, wurde das Gefäßsystem blau gefärbt. Der mutierte Embryo zeigt starke Wachstumsdefekte und ein zerstörtes Gefäßsystem. Dieses Ergebnis wurde in der Fachzeitschrift Blood publiziert [4].")
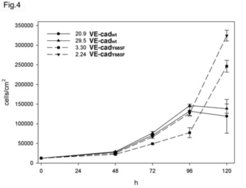
Wie VE-cadherin das Wachstum von Endothelzellen beschränken kann
Die zentrale Bedeutung von VE-cadherin für die Integrität endothelialer Zellkontakte ist der Grund, warum intensiv nach weiteren intrazellulären Bindungspartnern dieses Adhäsionsmoleküls gesucht wurde. Es gelang der Arbeitsgruppe, eine Kinase zu finden, die in der Tat an eine bestimmte Region im intrazellulären Teil von VE-cadherin bindet, wenn dieser Teil an einem bestimmten Tyrosinrest phophoryliert wird. Die Forscher konnten zeigen, dass die Bindung dieser Kinase, genannt „C-terminal Src Kinase“ (Csk), an VE-cadherin eine Rolle spielt bei der Inhibition des Wachstums von Endothelzellen (Abb. 4).
 wurden entweder mit dem normalen Zelladhäsionsmolekül VE-cadherin (VE-cad-wt) oder mit der punktmutierten Form (VE-cad-Y685F), in der das Tyrosin 685 durch Phenylalanin ersetzt wurde, transfiziert, . Jeweils zwei unterschiedliche Zellklone wurden für jede Transfektion untersucht. Das Bild zeigt eine Wachstumskurve der Zellen. Nur die mit normalem VE-cadherin transfizierten Zellen zeigen ein Abknicken der Wachstumskurve, während die mit mutiertem VE-cadherin transfizierten Zellen auch bei hoher Zelldichte ungehindert weiterwachsen. Diese Ergebnisse wurden im EMBO Journal publiziert [5].")